© World Health Organization 1999
Report of a WHO Consultation
Part 1: Diagnosis and Classification of Diabetes Mellitus
World Health Organization
Department of Noncommunicable Disease Surveillance
Geneva
Department of Noncommunicable Disease Surveillance
Geneva

Contents - Copyright - Authors
Contents
Copyright and Reproduction
Authors
COPYRIGHT AND REPRODUCTION
( Top of page ) (Contents list )
COPYRIGHT AND REPRODUCTION
The views expressed in documents by named authors are solely the responsibility of those authors.
COPYRIGHT AND REPRODUCTION
CONTENTS
( Top of page )
CONTENTS
- 1. Introduction
- 2. Definition and diagnostic criteria for diabetes mellitus and other categories of glucose intolerance
- 3. Classification
- 4. Clinical staging of diabetes mellitus and other categories of glucose tolerance
- 4.1 Diabetes mellitus
- 4.2 Impaired glucose regulation - Impaired Glucose Tolerance and Impaired Fasting Glycaemia (IFG)
- 4.3 Normoglycaemia
- 5. Aetiological types
- 6. Gestational Hyperglycaemia and Diabetes
- 7. Description of aetiological types
- 7.1 Type 1 (beta-cell destruction, usually leading to absolute insulin deficiency)
- 7.2 Type 2 (predominantly insulin resistance with relative insulin deficiency
or predominantly an insulin secretory defect with/without insulin resistance) - 7.3 Other Specific Types
- 7.3.1 Genetic defects of beta-cell function
- 7.3.2 Genetic defects in insulin action
- 7.3.3 Diseases of the exocrine pancreas
- 7.3.4 Endocrinopathies
- 7.3.5 Drug- or chemical-induced diabetes
- 7.3.6 Infections
- 7.3.7 Uncommon but specific forms of immune-mediated diabetes mellitus
- 7.3.8 Other genetic syndromes sometimes associated with diabetes
- 8. The Metabolic Syndrome
- References
- Annex 1 The Oral Glucose Tolerance Test
- Annex 2 Methods for measuring substances in blood and urine
( Top of 'Contents' )
CONTENTS
WHO CONSULTATION
( Top of page )
WHO CONSULTATION
Members
- KGMM Alberti, University of Newcastle upon Tyne, UK (Co-Chairman)
- P Aschner, ACD and Javerlana University, Bogota, Colombia
- J-P Assal, University Hospital, Geneva, Switzerland
- PH Bennett, NIDDK, Phoenix, AZ, USA
- L Groop, University of Lund, Malmö, Sweden
- J Jervell, Rikshospitalet, Oslo, Norway
- Y Kanazawa, Jichi Medical School, Omiya, Japan
- H Keen, Guy's Hospital and Medical School, London, UK
- R Klein, University of Wisconsin Medical School, Madison, WI, USA
- J-C Mbanya, Centre Hospitalier et Universitaire de Yaoundé, Cameroon
- D McCarty, International Diabetes Institute, Caulfield, Australia (Rapporteur)
- A Motala, University of Natal, Congella, South Africa
- Pan X-R, China-Japan Friendship Hospital, Beijing, China PR (deceased 8 July 1997)
- A Ramachandran, Diabetes Research Centre, Madras, India
- N Samad, Dow Medical College & Civil Hospital, Karachi, Pakistan
- N Unwin, University of Newcastle upon Tyne, UK (Rapporteur)
- P Vardi, Schneider Children's Centre, Petah-Tikvah, Israel
- PZ Zimmet, International Diabetes Institute, Caulfield, Australia (Co-Chairman)
Secretariat
- A Alwan, World Health Organization, Geneva, Switzerland
- H King, World Health Organization, Geneva, Switzerland
Observers
- M Berrens, Bayer, Germany
- R Kahn, American Diabetes Association, USA
- J Nolan, Institute for Diabetes Discovery, USA
- S Pramming, Novo Nordisk, Denmark
- RA Rizza, American Diabetes Association, USA
( Top of 'WHO Consultation' )
WHO CONSULTATION
INTRODUCTION
( Top of page ) ( Contents ) ( References )
1. INTRODUCTION
An American Diabetes Association (ADA) expert group was convened to discuss these issues. It published its recommendations in 1997 (4). WHO convened a Consultation on the same subject in London, United Kingdom, in December 1996. In general, the ADA and WHO groups reached similar conclusions.
The provisional report of the WHO Consultation (5) solicited comments which were considered in preparing the present report. Both the provisional and the present report were prepared by Professor K.G.M.M. Alberti and Professor P.Z. Zimmet on behalf of the members of the Consultation. The meeting was made possible by generous financial support from Bayer, UK; Bayer, Germany; Novo Nordisk, Copenhagen, Denmark; and The Institute for Diabetes Discovery, New Haven, USA
( Top of 'Introduction' )
INTRODUCTION
DEFINITION AND DIAGNOSTIC CRITERIA
( Top of page ) ( Contents ) ( References )
2. DEFINITION AND DIAGNOSTIC CRITERIA FOR DIABETES MELLITUS AND OTHER CATEGORIES OF GLUCOSE INTOLERANCE
2.1 Definition
Several pathogenetic processes are involved in the development of diabetes. These include processes which destroy the beta cells of the pancreas with consequent insulin deficiency, and others that result in resistance to insulin action. The abnormalities of carbohydrate, fat and protein metabolism are due to deficient action of insulin on target tissues resulting from insensitivity or lack of insulin.
2.2 Diagnosis and diagnostic criteria
2.2.1 Diagnosis
2.3 Diagnostic criteria
Figure 1: Unstandardized (casual, random) blood glucose values in the diagnosis of diabetes in mmol l-1 (mg dl-1). Taken from the 1985 WHO Study Group Report (3).
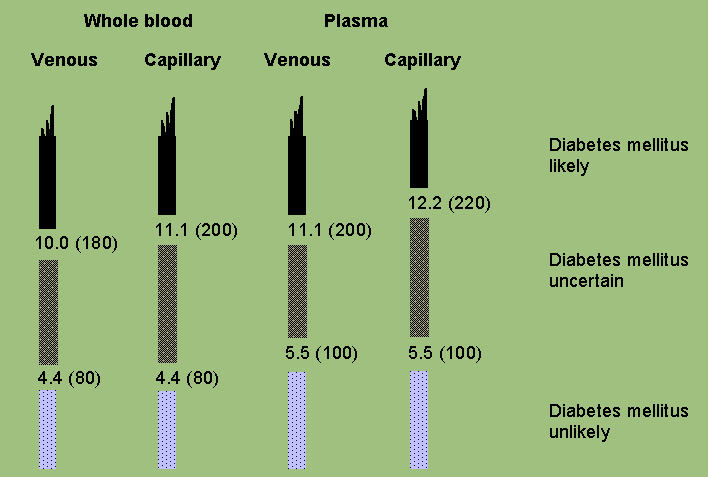
Table 1: Values for diagnosis of diabetes mellitus and other categories of hyperglycaemia
Glucose concentration, mmol l-1 (mg dl-1) | ||||
| Whole blood | Whole blood | Plasma* | ||
| Venous | Capillary | Venous | ||
| Diabetes Mellitus: | ||||
| Fasting | >=6.1 (>=110) | >=6.1 (>=110) | >=7.0 (>=126) | |
| or | ||||
| 2-h post glucose load | >=10.0 (>=180) | >=11.1 (>=200) | >=11.1 (>=200) | |
| or both | ||||
| Impaired Glucose Tolerance (IGT): | ||||
| Fasting (if measured) | <6.1 (<110) | <6.1 (<110) | <7.0 (<126) | |
| and | ||||
| 2-h post glucose load | >=6.7 (>=120) and | >=7.8 (>=140) and | >=7.8 (>=140) and | |
| <10.0 (<180) | <11.1 (<200) | <11.1 (<200) | ||
| Impaired Fasting Glycaemia (IFG): | ||||
| Fasting | >=5.6 (>=100) and | >=5.6 (>=100) and | >=6.1 (>=110) and | |
| <6.1 (<110) | <6.1 (<110) | <7.0 (<126) | ||
| and (if measured) | ||||
| 2-h post glucose load | <6.7 (<120) | <7.8 (<140) | <7.8 (<140) | |
For epidemiological or population screening purposes, the fasting or 2-h value after 75 g oral glucose may be used alone. For clinical purposes, the diagnosis of diabetes should always be confirmed by repeating the test on another day unless there is unequivocal hyperglycaemia with acute metabolic decompensation or obvious symptoms.
Glucose concentrations should not be determined on serum unless red cells are immediately removed, otherwise glycolysis will result in an unpredictable under-estimation of the true concentrations. It should be stressed that glucose preservatives do not totally prevent glycolysis. If whole blood is used, the sample should be kept at 0-4 °C or centrifuged immediately, or assayed immediately.
The new fasting criterion is chosen to represent a value which is at the upper end of the range that corresponds in diagnostic significance in many persons to that of the 2-h post-load concentration, which is not changed. This equivalence has been established from several population-based studies (6-8) and it also represents an optimal cut-off point to separate the components of bimodal frequency distributions of fasting plasma glucose concentrations seen in several populations. Furthermore, several studies have shown increased risk of microvascular disease in persons with fasting plasma glucose concentrations of 7.0 mmol l-1 (126 mg dl-1) and over (6), and of macrovascular disease in persons with such fasting concentrations, even in those with 2-h values of < 7.8 mmol l-1 (140 mg dl-1) (9). Nevertheless, in less obese subjects, in some ethnic groups and in the elderly lower fasting glucose levels may be seen in persons who have 2-h post-load glucose values that are diagnostic for diabetes.
( Top of 'Definition and Diagnostic Criteria' )
DEFINITION AND DIAGNOSTIC CRITERIA
CLASSIFICATION
( Top of page ) ( Contents ) ( References )
3. CLASSIFICATION
3.1 Earlier classifications
3.2 Revised classification
The clinical staging reflects that diabetes, regardless of its aetiology, progresses through several clinical stages during its natural history. Moreover, individual subjects may move from stage to stage in either direction. Persons who have, or who are developing, diabetes mellitus can be categorized by stage according to the clinical characteristics, even in the absence of information concerning the underlying aetiology. The classification by aetiological type results from improved understanding of the causes of diabetes mellitus.
All subjects with diabetes mellitus can be categorized according to clinical stage, and this is achievable in all circumstances. The stage of glycaemia may change over time depending on the extent of the underlying disease processes (Figure 2). The disease process may be present but may not have progressed far enough to cause hyperglycaemia. The aetiological classification reflects the fact that the defect or process which may lead to diabetes may be identifiable at any stage in the development of diabetes - even at the stage of normoglycaemia. Thus the presence of islet cell antibodies in a normoglycaemic individual makes it likely that that person has the Type 1 autoimmune process. Unfortunately there are few sensitive or highly specific indicators of the Type 2 process at present, although these are likely to be revealed as aetiology is more clearly defined. The same disease processes can cause impaired fasting glycaemia and/or impaired glucose tolerance without fulfilling the criteria for the diagnosis of diabetes mellitus. In some individuals with diabetes, adequate glycaemic control can be achieved with weight reduction, exercise and/or oral agents. These individuals, therefore, do not require insulin and may even revert to IGT or normoglycaemia. Other individuals require insulin for adequate glycaemic control but can survive without it. These individuals, by definition, have some residual insulin secretion. Individuals with extensive beta-cell destruction, and therefore no residual insulin secretion, require insulin for survival. The severity of the metabolic abnormality can either regress (e.g. with weight reduction), progress (e.g. with weight gain), or stay the same.
3.3 Terminology (Table 2)
- The terms Type 1 and Type 2 should be reintroduced. The aetiological type named Type 1 encompasses the majority of cases which are primarily due to pancreatic islet beta-cell destruction and are prone to ketoacidosis. Type 1 includes those cases attributable to an autoimmune process, as well as those with beta-cell destruction and who are prone to ketoacidosis for which neither an aetiology nor a pathogenesis is known (idiopathic). It does not include those forms of beta-cell destruction or failure to which specific causes can be assigned (e.g. cystic fibrosis, mitochondrial defects, etc.). Some subjects with this type can be identified at earlier clinical stages than "diabetes mellitus".
- The type named Type 2 includes the common major form of diabetes which results from defect(s) in insulin secretion, almost always with a major contribution from insulin resistance. It has been argued that a lean phenotype of Type 2 diabetes mellitus in adults found in the Indian sub-continent may be very distinct from the more characteristic form of Type 2 found in Caucasians. Not enough information is available, however, to characterize such subjects separately.
- A recent international workshop reviewed the evidence for, and characteristics of, diabetes mellitus seen in undernourished populations (16,17). Whilst it appears that malnutrition may influence the expression of several types of diabetes, the evidence that diabetes can be caused by malnutrition or protein deficiency per se is not convincing. Therefore, it is recommended that the class "Malnutrition-related diabetes" (MRDM) be deleted. The former subtype of MRDM, Protein-deficient Pancreatic Diabetes (PDPD or PDDM), may be considered as a malnutrition modulated or modified form of diabetes mellitus for which more studies are needed. The other former subtype of MRDM, Fibrocalculous Pancreatic Diabetes (FCPD), is now classified as a disease of the exocrine pancreas, fibrocalculous pancreatopathy, which may lead to diabetes mellitus.
- The class "Impaired Glucose Tolerance" is now classified as a stage of impaired glucose regulation, since it can be observed in any hyperglycaemic disorder, and is itself not diabetes.
- A clinical stage of Impaired Fasting Glycaemia has been introduced to classify individuals who have fasting glucose values above the normal range, but below those diagnostic of diabetes.
- Gestational Diabetes is retained but now encompasses the groups formerly classified as Gestational Impaired Glucose Tolerance (GIGT) and Gestational Diabetes Mellitus (GDM).
Table 2: Aetiological Classification of Disorders of Glycaemia*
- Type 1
- (beta-cell destruction, usually leading to absolute insulin deficiency)
- Autoimmune
- Idiopathic
- Type 2
- (may range from predominantly insulin resistance with relative insulin deficiency to a predominantly secretory defect with or without insulin resistance)
- Other specific types (see Table 3)
- Genetic defects of beta-cell function
- Genetic defects in insulin action
- Diseases of the exocrine pancreas
- Endocrinopathies
- Drug- or chemical-induced
- Infections
- Uncommon forms of immune-mediated diabetes
- Other genetic syndromes sometimes associated with diabetes
- Gestational diabetes**
**Includes the former categories of gestational impaired glucose tolerance and gestational diabetes.
( Top of 'Classification' )
CLASSIFICATION
CLINICAL STAGING
( Top of page ) ( Contents ) ( References )
4. CLINICAL STAGING OF DIABETES MELLITUS AND OTHER CATEGORIES OF GLUCOSE TOLERANCE
Figure 2: Disorders of glycaemia: aetiological types and clinical stages.
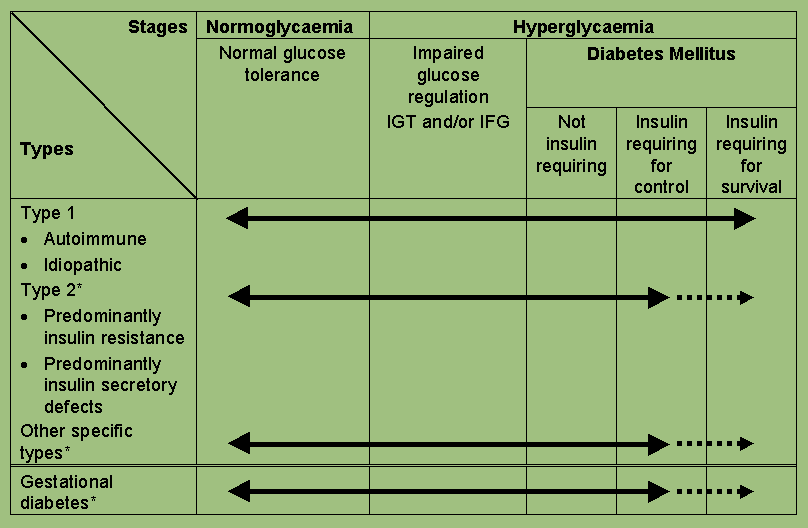
4.1 Diabetes mellitus
4.2 Impaired glucose regulation - Impaired Glucose Tolerance (IGT) and
Impaired Fasting Glycaemia (IFG)
IGT, rather than being a class as in the previous classification, is categorized as a stage in the natural history of disordered carbohydrate metabolism. A stage of IFG is also recognized because such subjects, like those with IGT, have increased risks of progressing to diabetes and macrovascular disease, although prospective data are sparse and early data suggest a lower risk of progression than IGT (18), although a similar CVD risk factor profile has been shown in IFG and IGT subjects (19). IFG refers to fasting glucose concentrations which are lower than those required to diagnose diabetes mellitus but higher than the "normal" reference range.
The values for IFG are a fasting plasma glucose concentration of 6.1 mmol l-1 (110 mg dl-1) or greater (whole blood 5.6 mmol l-1; 100 mg dl-1), but less than 7.0 mmol l-1 (126 mg dl-1) (whole blood 6.1 mmol l-1; 110 mg dl-1). If an OGTT is performed, some individuals with IFG will have IGT or diabetes, but this cannot be determined without an OGTT. If resources allow, it is recommended that all those with IFG have an OGTT to exclude the diagnosis of diabetes.
Individuals who meet criteria for IGT or IFG may be euglycaemic in their daily lives as shown by normal or near-normal glycated haemoglobin levels. IGT and IFG are not clinical entities in their own right, but rather risk categories for future diabetes and/or cardiovascular disease (20,21). They can occur as an intermediate stage in any of the disease processes listed in Table 2. IGT is often associated with the Metabolic Syndrome (Insulin Resistance Syndrome) (22). Thus, IGT may not be directly involved in the pathogenesis of cardiovascular disease, but rather may serve as an indicator or marker of enhanced risk by virtue of its correlation with the other elements of the Metabolic Syndrome that are cardiovascular risk factors. Self-evidently, those individuals with IGT manifest glucose intolerance only when challenged with an oral glucose load.
4.3 Normoglycaemia
The pathological or aetiological processes which often lead to diabetes mellitus begin, and may be recognizable, in some subjects who have normal glucose tolerance. Recognition of the pathological process at an early stage may be useful if progression to more advanced stages can be prevented. Conversely, effective treatments, or occasionally the natural history of some forms of diabetes mellitus, may result in reversion of hyperglycaemia to a state of normoglycaemia. The proposed classification includes a stage of normoglycaemia in which persons who have evidence of the pathological processes which may lead to diabetes mellitus, or in whom a reversal of the hyperglycaemia has occurred, are classified.
( Top of 'Clinical Staging' )
CLINICAL STAGING
AETIOLOGICAL TYPES
( Top of page ) ( Contents ) ( References )
5. AETIOLOGICAL TYPES (see also section 7. and Table 2)
5.1 Type 1
5.2 Type 2
5.3 Other specific types (Table 3)
( Top of 'Aetiological Types' )
AETIOLOGICAL TYPES
GESTATIONAL HYPERGLYCAEMIA AND DIABETES
( Top of page ) ( Contents ) ( References )
6. GESTATIONAL HYPERGLYCAEMIA AND DIABETES
Women who become pregnant and who are known to have diabetes mellitus which antedates pregnancy do not have gestational diabetes but have "diabetes mellitus and pregnancy" and should be treated accordingly before, during, and after the pregnancy.
In the early part of pregnancy (e.g. first trimester and first half of second trimester) fasting and postprandial glucose concentrations are normally lower than in normal, non-pregnant women. Elevated fasting or postprandial plasma glucose levels at this time in pregnancy may well reflect the presence of diabetes which has antedated pregnancy, but criteria for designating abnormally high glucose concentrations at this time have not yet been established. The occurrence of higher than usual plasma glucose levels at this time in pregnancy mandates careful management and may be an indication for carrying out an OGTT (Annex 1). Nevertheless, normal glucose tolerance in the early part of pregnancy does not itself establish that gestational diabetes may not develop later.
Individuals at high risk for gestational diabetes include older women, those with previous history of glucose intolerance, those with a history of large for gestational age babies, women from certain high-risk ethnic groups, and any pregnant woman who has elevated fasting, or casual, blood glucose levels. It may be appropriate to screen pregnant women belonging to high-risk populations during the first trimester of pregnancy in order to detect previously undiagnosed diabetes mellitus. Formal systematic testing for gestational diabetes is usually done between 24 and 28 weeks of gestation.
6.1 Diagnosis of gestational diabetes
( Top of 'Gestational Hyperglycaemia and Diabetes' )
GESTATIONAL HYPERGLYCAEMIA AND DIABETES
DESCRIPTION OF AETIOLOGICAL TYPES
( Top of page ) ( Contents ) ( References )
7. DESCRIPTION OF AETIOLOGICAL TYPES
7.1 Type 1 (beta-cell destruction, usually leading to absolute insulin deficiency)
7.1.1 Autoimmune Diabetes Mellitus
Markers of immune destruction, including islet cell autoantibodies, and/or autoantibodies to insulin, and autoantibodies to glutamic acid decarboxylase (GAD) are present in 85-90 % of individuals with Type 1 diabetes mellitus when fasting diabetic hyperglycaemia is initially detected (30). The peak incidence of this form of Type 1 diabetes occurs in childhood and adolescence, but the onset may occur at any age, ranging from childhood to the ninth decade of life (31). There is a genetic predisposition to autoimmune destruction of beta cells, and it is also related to environmental factors that are still poorly defined. Although patients are usually not obese when they present with this type of diabetes, the presence of obesity is not incompatible with the diagnosis. These patients may also have other autoimmune disorders such as Graves' disease, Hashimoto's thyroiditis, and Addison's disease (32).
7.2 Type 2 (predominantly insulin resistance with relative insulin deficiency or predominantly an insulin secretory defect with/without insulin resistance)
The majority of patients with this form of diabetes are obese, and obesity itself causes or aggravates insulin resistance (39,40). Many of those who are not obese by traditional weight criteria may have an increased percentage of body fat distributed predominantly in the abdominal region (41). Ketoacidosis is infrequent in this type of diabetes; when seen it usually arises in association with the stress of another illness such as infection (42,43). Whereas patients with this form of diabetes may have insulin levels that appear normal or elevated, the high blood glucose levels in these diabetic patients would be expected to result in even higher insulin values had their beta-cell function been normal (44). Thus, insulin secretion is defective and insufficient to compensate for the insulin resistance. On the other hand, some individuals have essentially normal insulin action, but markedly impaired insulin secretion. Insulin sensitivity may be increased by weight reduction, increased physical activity, and/or pharmacological treatment of hyperglycaemia but is not restored to normal (45,46). The risk of developing Type 2 diabetes increases with age, obesity, and lack of physical activity (47,48). It occurs more frequently in women with prior GDM and in individuals with hypertension or dyslipidaemia. Its frequency varies in different racial/ethnic subgroups (47-50). It is often associated with strong familial, likely genetic, predisposition (49-51). However, the genetics of this form of diabetes are complex and not clearly defined.
Some patients who present with a clinical picture consistent with Type 2 diabetes have autoantibodies similar to those found in Type 1 diabetes, and may masquerade as Type 2 diabetes if antibody determinations are not made. Patients who are non-obese or who have relatives with Type 1 diabetes and who are of Northern European origin may be suspected of having late onset Type 1 diabetes.
7.3 Other Specific Types
Table 3: Other Specific Types of Diabetes
- Chromosome 20, HNF4alpha (MODY1)
- Chromosome 7, glucokinase (MODY2)
- Chromosome 12, HNF1alpha (MODY3)
- Chromosome 13, IPF-1 (MODY4)
- Mitochondrial DNA 3243 mutation
- Others
- Type A insulin resistance
- Leprechaunism
- Rabson-Mendenhall syndrome
- Lipoatrophic diabetes
- Others
- Fibrocalculous pancreatopathy
- Pancreatitis
- Trauma / pancreatectomy
- Neoplasia
- Cystic fibrosis
- Haemochromatosis
- Others
- Cushing's syndrome
- Acromegaly
- Phaeochromocytoma
- Glucagonoma
- Hyperthyroidism
- Somatostatinoma
- Others
Infections
- Congenital rubella
- Cytomegalovirus
- Others
- Insulin autoimmune syndrome (antibodies to insulin)
- Anti-insulin receptor antibodies
- "Stiff Man" syndrome
- Others
Point mutations in mitochondrial DNA have been found to be associated with diabetes mellitus and deafness (59). The most common mutation occurs at position 3243 in the tRNA leucine gene, leading to an A to G substitution. An identical lesion occurs in the MELAS syndrome (Mitochondrial myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like syndrome); however, diabetes is not part of this syndrome, suggesting for unknown reasons different phenotypic expressions of this genetic lesion (60).
Genetic abnormalities that result in the inability to convert proinsulin to insulin have been identified in a few families. Such traits are usually inherited in an autosomal dominant pattern (61,62) and the resultant carbohydrate intolerance is mild. Similarly, mutant insulin molecules with impaired receptor binding have been identified in a few families. These are also associated with autosomal inheritance and either normal or only mildly impaired carbohydrate metabolism (63,64).
Somatostatinoma, and aldosteronoma-induced hypokalaemia, can cause diabetes, at least in part by inhibiting insulin secretion (74,75). Hyperglycaemia generally resolves following successful removal of the tumour.
Table 4: Drug- or Chemical-induced Diabetes
- Nicotinic acid
- Glucocorticoids
- Thyroid hormone
- Alpha-adrenergic agonists
- Beta-adrenergic agonists
- Thiazides
- Dilantin
- Pentamidine
- Vacor
- Interferon-alpha therapy
- Others
Anti-insulin receptor antibodies can cause diabetes by binding to the insulin receptor thereby reducing the binding of insulin to target tissues (89). However, these antibodies also can act as an insulin agonist after binding to the receptor and can thereby cause hypoglycaemia (90). Anti-insulin receptor antibodies are occasionally found in patients with systemic lupus erythematosus and other autoimmune diseases (91). As in other states of extreme insulin resistance, patients with anti-insulin receptor antibodies often have acanthosis nigricans. In the past, this syndrome was termed Type B insulin resistance.
Table 5: Other Genetic Syndromes Sometimes Associated with Diabetes
- Down's syndrome
- Friedreich's ataxia
- Huntington's chorea
- Klinefelter's syndrome
- Lawrence-Moon-Biedel syndrome
- Myotonic dystrophy
- Porphyria
- Prader-Willi syndrome
- Turner's syndrome
- Wolfram's syndrome
- Others
( Top of 'Description of Aetiological Types' )
DESCRIPTION OF AETIOLOGICAL TYPES
METABOLIC SYNDROME
( Top of page ) ( Contents ) ( References )
8. THE METABOLIC SYNDROME
Often a person with abnormal glucose tolerance (IGT or diabetes) will be found to have at least one or more of the other cardiovascular disease (CVD) risk components (22). This clustering has been labelled variously as Syndrome X (22), the Insulin Resistance Syndrome (47), or the Metabolic Syndrome (47).
Epidemiological studies confirm that this syndrome occurs commonly in a wide variety of ethnic groups including Caucasians, Afro-Americans, Mexican-Americans, Asian Indians, Chinese, Australian Aborigines, Polynesians and Micronesians (47,93). In 1988 Reaven focused attention on this cluster, naming it Syndrome X (22). Central obesity was not included in the original description so the term Metabolic Syndrome is now favoured.
Evidence is accumulating that insulin resistance may be the common aetiological factor for the individual components of the Metabolic Syndrome (47,93,94), although there appears to be heterogeneity in the strength of the insulin resistance relationship with different components between, and even within, populations. Alone, each component of the cluster conveys increased CVD risk, but as a combination they become much more powerful (95). This means that the management of persons with hyperglycaemia and other features of the Metabolic Syndrome should focus not only on blood glucose control but also include strategies for reduction of the other CVD risk factors (96).
It is well documented that the features of the Metabolic Syndrome can be present for up to 10 years before detection of the glycaemic disorders (97). This is important in relation to the aetiology of the hyperglycaemia and the associated CVD risk, and the potential to prevent CVD and its morbidity and mortality in persons with glucose intolerance.
The Metabolic Syndrome with normal glucose tolerance identifies the subject as a member of a group at very high risk of future diabetes. Thus, vigorous early management of the syndrome may have a significant impact on the prevention of both diabetes and cardiovascular disease (98).
8.1 Definition
- Impaired glucose regulation or diabetes (see Table 1)
- Insulin resistance (under hyperinsulinaemic, euglycaemic conditions, glucose uptake below lowest quartile for background population under investigation)
- Raised arterial pressure >= 140/90 mmHg
- Raised plasma triglycerides (>= 1.7 mmol l-1; 150 mg dl-1) and/or low HDL-cholesterol (< 0.9 mmol l-1, 35 mg dl-1 men; < 1.0 mmol l-1, 39 mg dl-1 women)
- Central obesity (males: waist to hip ratio > 0.90; females: waist to hip ratio > 0.85) and/or BMI > 30 kg m-2
- Microalbuminuria (urinary albumin excretion rate >= 20 µg min-1 or albumin:creatinine ratio >= 30 mg g-1)
8.2 Future needs
( Top of 'Metabolic Syndrome' )
METABOLIC SYNDROME
REFERENCES
( Top of page ) ( Contents )
REFERENCES
1. WHO Expert Committee on Diabetes Mellitus. Second Report. Geneva: WHO, 1980. Technical Report Series 646.
2. National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes 1979; 28: 1039-57.
3. World Health Organization. Diabetes Mellitus: Report of a WHO Study Group. Geneva: WHO, 1985. Technical Report Series 727.
4. The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 1997; 20: 1183-97.
5. Alberti KGMM, Zimmet PZ for the WHO Consultation. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. Diabetic Medicine 1998; 15: 539-553.
6. McCance DR, Hanson RL, Charles MA, Jacobsson LTH, Pettitt DJ, Bennett PH et al. Comparison of tests for glycated haemoglobin and fasting and two hour plasma glucose concentrations as diagnostic methods for diabetes. BMJ 1994; 308: 1323-28.
7. Finch CF, Zimmet PZ, Alberti KGMM. Determining diabetes prevalence: a rational basis for the use of fasting plasma glucose concentrations? Diabetic Medicine 1990; 7: 603-10.
8. Engelgau MM, Thompson TJ, Herman WH, Boyle JP, Aubert RE, Kenny SJ et al. Comparison of fasting and 2-hour glucose and HbA1c levels for diagnosing diabetes: diagnostic criteria and performance revisited. Diabetes Care 1997; 20: 785-91.
9. Charles MA, Balkau B, Vauzelle-Kervoeden F, Thibult N, Eschwège E. Revision of diagnostic criteria for diabetes (Letter). Lancet 1996; 348: 1657-58.
10. DECODE Study Group on behalf of the European Diabetes Epidemiology Study Group. Will new diagnostic criteria for diabetes mellitus change phenotype of patients with diabetes? Reanalysis of European epidemiological data. BMJ 1998; 317: 371-375.
11. De Vegt F, Dekker JM, Stehouwer CDA, Nijpels G, Bouter LM, Heine RJ. The 1997 American Diabetes Association criteria versus the 1985 World Health Organization criteria for the diagnosis of abnormal glucose tolerance: poor agreement in the Hoorn Study. Diabetes Care 1998; 21: 1686-1690.
12. Wahl PW, Savage PJ, Psaty BM, Orchard TJ, Robbins JA, Tracy RP. Diabetes in older adults: comparison of 1997 American Diabetes Association classification of diabetes mellitus with 1985 WHO classification. Lancet 1998; 352: 1012-1015.
13. Harris MI, Eastman RC, Cowie CC, Flegal KM, Eberhardt MS. Comparison of diabetes diagnostic categories in the US population according to 1997 American Diabetes Association and 1980-1985 World Health Organization diagnostic criteria. Diabetes Care 1997; 20: 1859-62.
14. Ramachandran A, Snehalatha C, Latha E, Vijay V. Evaluation of the use of fasting plasma glucose as a new diagnostic criterion for diabetes in Asian Indian population (Letter). Diabetes Care 1998; 21: 666-67.
15. Kuzuya T, Matsuda A. Classification of diabetes on the basis of etiologies versus degree of insulin deficiency. Diabetes Care 1997; 20: 219-20.
16. Hoet JJ, Tripathy BB, Rao RH, Yajnik CS. Malnutrition and diabetes in the tropics. Diabetes Care 1996; 19: 1014-17.
17. Tripathy BB, Samal KC. Overview and consensus statement on diabetes in tropical areas. Diabetes Metab Rev 1997; 13: 63-76.
18. Shaw JE, Zimmet PZ, de Courten M, Dowse GK, Gareeboo H, Hemraj F et al. IFG or IGT: what best predicts future diabetes? A view of the new ADA recommendations. Diabetes Care 1999 (In press)
19. Larsson H, Berglund G, Lindgärde F, Ahrén B. Comparison of ADA and WHO criteria for diagnosis of diabetes and glucose intolerance. Diabetologia 1998; 41: 1124-1125.
20. Fuller JH, Shipley MJ, Rose G, Jarrett RJ, Keen H. Coronary heart disease risk and impaired glucose tolerance: the Whitehall Study. Lancet 1980; i: 1373-76.
21. Alberti KGMM. The clinical implications of Impaired Glucose Tolerance. Diabet Med 1996; 13: 927-37.
22. Reaven GM. Role of insulin resistance in human disease. Diabetes 1988; 37: 1595-607.
23. McCance DR, Hanson RL, Pettitt DJ, Bennett PH, Hadden DR, Knowler WC. Diagnosing diabetes mellitus - do we need new criteria? Diabetologia 1997; 40: 247-55.
24. Zimmet PZ, Tuomi T, Mackay R, Rowley MJ, Knowles W, Cohen M et al. Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabetic Med 1994; 11: 299-303.
25. Humphrey ARG, McCarty DJ, Mackay IR, Rowley MJ, Dwyer T, Zimmet P. Autoantibodies to glutamic acid decarboxylase and phenotypic features associated with early insulin treatment in individuals with adult-onset diabetes mellitus. Diabetic Med 1998; 15: 113-19.
26. Japan and Pittsburgh Childhood Diabetes Research Groups. Coma at onset of young insulin-dependent diabetes in Japan: the result of a nationwide survey. Diabetes 1985; 34: 1241-46.
27. Zimmet PZ. The pathogenesis and prevention of diabetes in adults. Diabetes Care 1995; 18: 1050-64.
28. Willis JA, Scott RS, Brown LJ, Forbes LV, Schmidli RS, Zimmet PZ et al. Islet cell antibodies and antibodies against glutamic acid decarboxylase in newly diagnosed adult-onset diabetes mellitus. Diabetes Res Clin Pract 1996; 33: 89-97.
29. Hother-Nielsen O, Faber O, Sørensen NS, Beck-Nielsen H. Classification of newly diagnosed diabetic patients as insulin-requiring or non-insulin-requiring based on clinical and biochemical variables. Diabetes Care 1988; 11: 531-37.
30. Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA et al. Predicting type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996; 45: 926-33.
31. Mølbak AG, Christau B, Marner B, Borch-Johnsen K, Nerup J. Incidence of insulin-dependent diabetes mellitus in age groups over 30 years in Denmark. Diabet Med 1994; 11: 650-55.
32. Betterle C, Zanette F, Pedini B, Presotto F, Rapp LB, Monsciotti CM et al. Clinical and subclinical organ-specific autoimmune manifestations in type 1 (insulin-dependent) diabetic patients and their first-degree relatives. Diabetologia 1983; 26: 431-36.
33. McLarty DG, Athaide I, Bottazzo GF, Swai ABM, Alberti KGMM. Islet cell antibodies are not specifically associated with insulin-dependent diabetes in rural Tanzanian Africans. Diabetes Res Clin Pract 1990; 9: 219-24.
34. Ahrén B, Corrigan CB. Intermittent need for insulin in a subgroup of diabetic patients in Tanzania. Diabet Med 1984; 2: 262-64.
35. DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 635-712.
36. Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E et al. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes. Prospective Study of Pima Indians. N Engl J Med 1993; 329: 1988-92.
37. Mooy JM, Grootenhuis PA, de Vries H, Valkenburg HA, Bouter LM, Kostense PJ et al. Prevalence and determinants of glucose intolerance in a Dutch population. The Hoorn Study. Diabetes Care 1995; 18: 1270-73.
38. Harris MI. Undiagnosed NIDDM; clinical and public health issues. Diabetes Care 1993; 16: 642-52.
39. Campbell PJ, Carlson MG. Impact of obesity on insulin action in NIDDM. Diabetes 1993; 42: 405-10.
40. Bogardus C, Lillioja S, Mott DM, Hollenbeck C, Reaven G. Relationship between degree of obesity and in vivo insulin action in man. Am J Physiol 1985; 248: E286-E291.
41. Kissebah AH, Vydelingum N, Murray R, Evans DF, Hartz AJ, Kalkhoff RK et al. Relationship of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab 1982; 54: 254-60.
42. Banerji MA, Chaiken RI, Huey H, Tuomi T, Norin AJ, MacKay IR et al. GAD antibody negative NIDDM in adult black subjects with diabetic ketoacidosis and increased frequency of human leukocyte antigen DR3 and DR4: flatbush diabetes. Diabetes 1994; 43: 741-45.
43. Umpierrez GE, Casals MMC, Gebhardt SSP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44: 790-95.
44. Polonsky KS, Sturis J, Bell GI. Non-insulin-dependent diabetes mellitus: a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med 1996; 334: 777-84.
45. Simonson DC, Ferrannini E, Bevilacqua S, Smith D, Barrett E, Carlson R et al. Mechanism of improvement in glucose metabolism after chronic glyburide therapy. Diabetes 1984; 33: 838-45.
46. Wing RR, Blair EH, Bononi P, Marcus MD, Watanabe R, Bergman RN. Caloric restriction per se is a significant factor in improvements in glycemic control and insulin sensitivity during weight loss in obese NIDDM patients. Diabetes Care 1994; 17: 30-36.
47. Zimmet PZ. Kelly West Lecture 1991: challenges in diabetes epidemiology: from West to the rest. Diabetes Care 1992; 15: 232-52.
48. Harris MI, Cowie CC, Stern MP, Boyko ES, Reiber GE, Bennett PH, eds. Diabetes in America. 2nd edn. Washington DC: US Government Printing Office, 1995 (NIH publ. No. 95-1468).
49. Valle T, Tuomilehto J, Eriksson J. Epidemiology of NIDDM in Europids. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 125-42.
50. de Courten M, Bennett PH, Tuomilehto J, Zimmet P. Epidemiology of NIDDM in Non-Europids. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 143-70.
51. Knowler WC, Nelson RG, Saad M, Bennett PH, Pettitt DJ. Determinants of diabetes mellitus in the Pima Indians. Diabetes Care 1993; 16: 216-27.
52. Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ et al. Altered insulin secretory response to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY 3 on chromosome 20. Diabetes 1996; 45: 1503-10.
53. Clement K, Pueyo ME, Vaxillaire M, Rakotoambinina B, Thuillier F, Passa P et al. Assessment of insulin sensitivity in glucokinase-deficient subjects. Diabetologia 1996; 39: 82-90.
54. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY 3). Nature 1996; 384: 455-58.
55. Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes. Nature 1992; 356: 162-64.
56. Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes. Nature 1992; 356: 721-22.
57. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY 1). Nature 1996; 384: 458-60.
58. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nature Genetics 1997; 117: 138-39.
59. Walker M, Turnbull DM. Mitochondrial related diabetes: a clinical perspective. Diabet Med 1997; 14: 1007-09.
60. Johns DR. Mitochondrial DNA and disease. N Engl J Med 1995; 333: 638-44.
61. Gruppuso PA, Gorden P, Kahn CR, Cornblath M, Zeller WP, Schwartz R. Familial hyperproinsulinemia due to a proposed defect in conversion of proinsulin to insulin. N Engl J Med 1984; 311: 629-34.
62. Robbins DC, Shoelson SE, Rubenstein AH, Tager HS. Familial hyperproinsulinemia: two cohorts secreting indistinguishable type II intermediates of proinsulin conversion. J Clin Invest 1984; 73: 714-19.
63. Haneda M, Polonsky KS, Bergenstal RM, Jaspan JB, Shoelson SE, Blix PM et al. Familial hyperinsulinemia due to a structurally abnormal insulin. Definition of an emerging new clinical syndrome. N Engl J Med 1984; 310: 1288-94.
64. Sanz N, Karam JH, Horita S, Bell GI. Prevalence of insulin-gene mutations in non-insulin-dependent diabetes mellitus. N Engl J Med 1986; 314: 1322-23.
65. Kahn CR, Flier JS, Bar RS, Archer JA, Gorden P, Martin MM et al. The syndromes of insulin resistance and acanthosis nigricans. N Engl J Med 1976; 294: 739-45.
66. Taylor SI. Lilly Lecture: molecular mechanisms of insulin resistance: lessons from patients with mutations in the insulin-receptor gene. Diabetes 1992; 41: 1473-90.
67. Gullo L, Pezzilli R, Morselli-Labate AM, and the Italian Pancreatic Cancer Study Group. Diabetes and the risk of pancreatic cancer. N Engl J Med 1994; 331: 81-84.
68. Larsen S, Hilsted J, Tronier B, Worning H. Metabolic control and B cell function in patients with insulin-dependent diabetes mellitus secondary to chronic pancreatitis. Metabolism 1987; 36: 964-67.
69. Permert J, Larsson J, Westermark GT, Herrington MK, Christmanson L, Pour PM et al. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med 1994; 330: 313-18.
70. Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS et al. Insulin sensitivity in cystic fibrosis. Diabetes 1994; 43: 1020-26.
71. Phelps G, Chapman I, Hall P, Braund W, Mackinnon M. Prevalence of genetic haemochromatosis among diabetic patients. Lancet 1989; ii: 233-34.
72. Yajnik CS, Shelgikar KM, Naik SS, Kanitkar SV, Orskov H, Alberti KGMM et al. The ketoacidosis-resistance in fibro-calculous-pancreatic-diabetes. Diabetes Res Clin Pract 1992; 15: 149-56.
73. MacFarlane IA. Endocrine diseases and diabetes mellitus. In: Pickup JC, Williams G, eds. Textbook of Diabetes. 2nd edn. Oxford: Blackwell, 1997: pp 64.1-64.20.
74. Krejs GJ, Orci L, Conlon JM, Ravazzola M, Davis GR, Raskin P et al. Somatostatinoma syndrome. N Engl J Med 1979; 301: 285-92.
75. Conn JW. Hypertension, the potassium ion and impaired carbohydrate tolerance. N Engl J Med 1965; 273: 1135-43.
76. Pandit MK, Burke J, Gustafson AB, Minocha A, Peiris AN. Drug-induced disorders of glucose tolerance. Ann Intern Med 1993; 118: 529-40.
77. O'Byrne S, Feely J. Effects of drugs on glucose tolerance in non-insulin-dependent diabetes (parts I and II). Drugs 1990; 40: 203-19.
78. Gallanosa AG, Spyker DA, Curnow RT. Diabetes mellitus associated with autonomic and peripheral neuropathy after Vacor poisoning: a review. Clin Toxicol 1981; 18: 441-49.
79. Esposti MD, Ngo A, Myers MA. Inhibition of mitochondrial complex I may account for IDDM induced by intoxication with rodenticide Vacor. Diabetes 1996; 45: 1531-34.
80. Assan R, Perronne C, Assan D, Chotard L, Mayaud C, Matheron S et al. Pentamidine-induced derangements of glucose homeostasis. Diabetes Care 1995; 18: 47-55.
81. Forrest JA, Menser MA, Burgess JA. High frequency of diabetes mellitus in young patients with congenital rubella. Lancet 1971; ii: 332-34.
82. King ML, Bidwell D, Shaikh A, Voller A, Banatvala JE. Coxsackie-B-virus-specific IgM responses in children with insulin-dependent (juvenile-onset; type 1) diabetes mellitus. Lancet 1983; i: 1397-99.
83. Karjalainen J, Knip M, Hyoty H, Linikki P, Ilonen J, Kaar M-L et al. Relationship between serum insulin antibodies, islet cell antibodies and Coxsackie-B4 and mumps virus-specific antibodies at the clinical manifestation of type 1 (insulin-dependent) diabetes. Diabetologia 1988; 31: 146-52.
84. Pak CY, Eun H, McArthur RG, Yoon J. Association of cytomegalovirus infection with autoimmune type 1 diabetes. Lancet 1988; ii: 1-4.
85. Hirata Y, Ishizu H, Ouchi N et al. Insulin autoimmunity in a case of spontaneous hypoglycaemia. J Jpn Diabet Soc 1970; 13: 312-20.
86. Bodansky HJ, Grant PJ, Dean BM, McNally J, Bottazzo GF, Hambling MH et al. Islet-cell antibodies and insulin autoantibodies in association with common viral infections. Lancet 1986; ii: 1351-53.
87. Solimena M, De Camilli P. Autoimmunity to glutamic acid decarboxylase (GAD) in Stiff-Man syndrome and insulin-dependent diabetes mellitus. Trends Neurosci 1991; 14: 452-57.
88. Fabris P, Betterle C, Floreani A, Greggio NA, de Lazzari F, Naccarato R et al. Development of type 1 diabetes mellitus during interferon alfa therapy for chronic HCV hepatitis (Letter). Lancet 1992; 340: 548.
89. Flier JS. Lilly Lecture: syndromes of insulin resistance: from patient to gene and back again. Diabetes 1992; 41: 1207-19.
90. Kahn CR, Baird KL, Flier JS, Jarrett DB. Effects of autoantibodies to the insulin receptor on isolated adipocytes. J Clin Invest 1977; 60: 1094-106.
91. Tsokos GC, Gorden P, Antonovych T, Wilson CB, Balow JE. Lupus nephritis and other autoimmune features in patients with diabetes mellitus due to autoantibody to insulin receptors. Ann Intern Med 1985; 102: 176-81.
92. Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995; 346: 1458-63.
93. Stern MP. The insulin resistance syndrome. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 255-83.
94. Haffner SM, Valdez RA, Hazuda HP, Mitchell BD, Morales PA, Stern MP. Prospective analysis of the insulin resistance syndrome (Syndrome X). Diabetes 1992; 41: 715-22.
95. Kaplan NM. The deadly quartet: upper body adiposity, glucose intolerance, hypertriglyceridaemia and hypertension. Arch Intern Med 1989; 149: 1514-20.
96. European Arterial Risk Policy Group on behalf of the International Diabetes Federation (European Region). A strategy for arterial risk assessment and management in Type 2 (non-insulin-dependent) diabetes. Diabet Med 1997; 14: 611-21.
97. Mykkänen L, Kuusisto J, Pyörälä K, Laakso M. Cardiovascular disease risk factors as predictors of Type 2 (non-insulin-dependent) diabetes mellitus in elderly subjects. Diabetologia 1993; 36: 553-59.
98. Eriksson KF, Lindegärde F. Prevention of Type 2 (non-insulin-dependent) diabetes mellitus by diet and physical exercise. Diabetologia 1991; 34: 891-98.
No comments:
Post a Comment